Abstract 1788: Mechanism of resistance to c-Met tyrosine kinase inhibitors in human melanoma
http://cancerres.aacrjournals.or ... etingAbstracts/1788
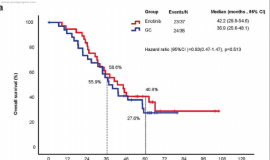
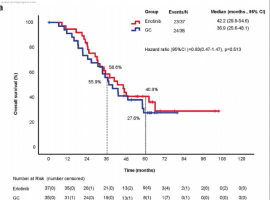
The over-expression of c-Met and activation by its ligand, hepatocyte growth factor (HGF), are known to cause significant tumor development and metastasis in human melanoma. Hence, c-Met is an attractive target for molecular therapeutic inhibition. To assess c-Met inhibition, we studied the effects of c-Met tyrosine kinase inhibitors (TKIs) in RU melanoma cells and found that JNJ38877605 and SU11274 (c-Met TKIs) inhibited cell growth in RU cells with an IC50 of 0.5 and 1µM, respectively. We then tested the therapeutic effects of JNJ38877605 in vivo. Five million RU melanoma cells were injected subcutaneously into the hind flanks of nude mice. Tumors were allowed to develop for a week after which daily oral treatments of 20 mg/kg JNJ38877605 or vehicle were given. We found that JNJ38877605 decreased the size of RU tumor xenografts in nude mice by 6 fold compared to mice treated with a vehicle control. Unfortunately, while c-Met inhibitors have been successful in clinical trials, acquired resistance to c-Met TKIs (ARQ197) and antibodies (MetMab) has been observed in cancer patients, resulting in tumor recurrence or lack of tumor CR. To determine the mechanism of c-Met TKI resistance, two melanoma cell line models, MU and RU, were exposed to increasing concentrations of SU11274. They proliferated in 7-10 fold higher concentrations of SU11274, and were cross resistant to 12 fold higher concentrations of JNJ38877605. After generating resistant cells, we measured HGF production and the expression levels of p-c-Met (Y1003) and p-c-Met (Y1234/1235) by immunoblotting to compare parental and resistant cells. We found that MU resistant cells exhibited upregulation of p-c-Met (Y1003) (6 fold) and p-c-met (Y1234/1235) (4 fold) compared to parental cells after treatment with HGF. Additionally, p-c-Met (Y1234/1235) was stable for only 30 min in parental cells compared to 60 min in resistant cells, where it was upregulated by 3-fold. This might be due to defects in c-Met ubiquitination resulting in its being recycled back or stabilized onto the membrane. Interestingly, we found that RU parental cells secreted 2 fold more HGF than the resistant cells. This indicates that resistant RU cells may not be as dependent on HGF induced activation in comparison to parental cells. In MU and RU resistant cells, we also found upregulation of p-mTOR (2 and 8 fold, respectively). Upregulation of phospho-pS6kinase (2 fold) and p-4E-BP1 (4 fold), downstream targets of p-mTOR, were also seen in MU cells. MU resistant cells also expressed increases in active β-catenin (protein downstream of Wnt signaling pathway) by 5 fold under no HGF stimulation and 2 fold after 60 min of HGF treatment. Our data indicates that the oral c-Met TKI, JNJ38877605, could be a promising therapeutic option for treating HGF producing melanoma tumors, but its effects could be further enhanced by the addition of mTOR and/or Wnt inhibitors to counteract acquired c-Met TKI resistance.
|
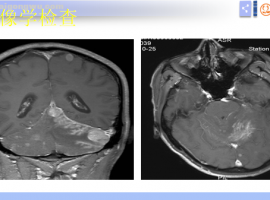 脑膜转移诊疗潘振宇教授科普总结
脑膜转移诊疗潘振宇教授科普总结
脑膜转移 中位治疗生存期 3-6月
脑膜转移的临床表现
脑膜转移诊疗潘振宇教授科普总结
脑膜转移诊疗潘振宇教授科普总结
脑膜转移 中位治疗生存期 3-6月
脑膜转移的临床表现
 母亲肺腺癌,9年靶向耐药后,如何选
2017年5月,因持续干咳入院检查,肺部弥漫型结节,支气管镜确诊肺腺癌,四期。基因检
母亲肺腺癌,9年靶向耐药后,如何选
2017年5月,因持续干咳入院检查,肺部弥漫型结节,支气管镜确诊肺腺癌,四期。基因检
 肺癌术前新辅助治疗,靶向治疗能否替
作者:闵
手术是广大病友眼中治愈癌症的神兵利器,可惜有一部分病友尽管处于中早期,
肺癌术前新辅助治疗,靶向治疗能否替
作者:闵
手术是广大病友眼中治愈癌症的神兵利器,可惜有一部分病友尽管处于中早期,
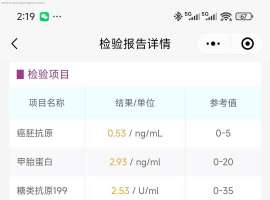
 请教下 3a 术后 检查
请教下 3a 术后 检查快两年了, 验血 不是每次都是 肿标 5项吗,我的复查 好像 有时
请教下 3a 术后 检查
请教下 3a 术后 检查快两年了, 验血 不是每次都是 肿标 5项吗,我的复查 好像 有时
 求助:1a1实体型低分化alk突变复发概
背景:35岁男性,无吸烟史,有熬夜习惯,检查前曾参与新房装修。24年7月底体检第一次
求助:1a1实体型低分化alk突变复发概
背景:35岁男性,无吸烟史,有熬夜习惯,检查前曾参与新房装修。24年7月底体检第一次






 显身卡
显身卡